Galaxy-P 101 - Building Up and Using a Proteomics Workflow¶
This section is heavily inspired by the Galaxy 101 maintained at Penn State by the core Galaxy team <http://wiki.galaxyproject.org/GalaxyTeam>.
In this walkthrough we will introduce you to basics of Galaxy-P:
- Uploading database using Protein Database Downloader.
- Getting raw data from an external source.
- Peaklist processing including options and parameters.
- X! tandem search.
- PeptideProphet processing of X!tandem search.
- Converting PepXML to a table.
- Using ProteinProphet to process PeptideProphet results.
- FDR analysis.
- Performing simple data manipulation.
- Understanding Galaxy’s history system.
- Creating and editing workflows.
- Applying workflows to your data.
What Are We Trying to Do?¶
We are trying to set up a search of one RAW file (your experimental data acquired on an LTQ/Orbitrap instrument) against a database using a search algorithm named X!tandem. The idea is to estimate number of valid matches by using PeptideProphet or ProteinProphet and FDR analysis. Once we have created this workflow we can apply this workflow to another dataset and get results. So let’s try it...
Setting Up Your Galaxy-P Account¶
Go to the User menu at the top of Galaxy-P interface and choose Register:

Follow the instructions as prompted.
Organizing Your Windows¶
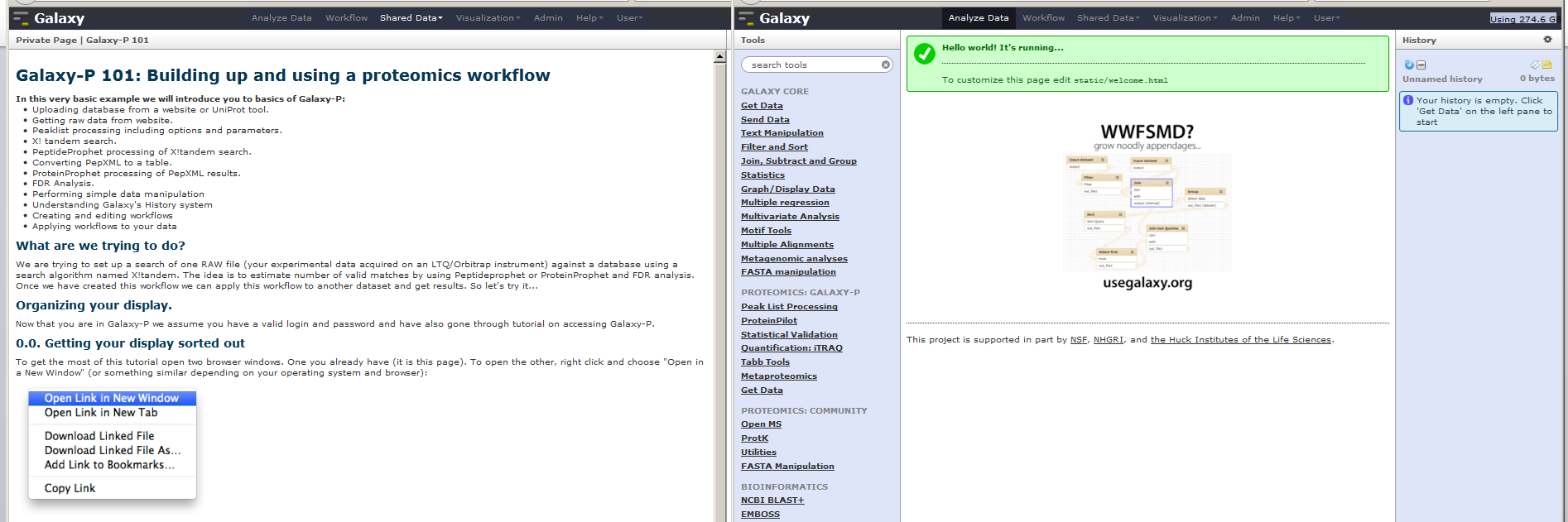
To get the most of this tutorial open two browser windows. One you already have (it is this page). To open the other, right click and choose “Open in a New Window” (or something similar depending on your operating system and browser):
Then organize your windows as something like this (depending on the size of your monitor you may or may not be able to organize things this way):
Loading a Search Database¶
There are a many options for how you can upload your search database (FASTA file with protein sequences). Three among these are:
- Protein Database Downloader.
- Use website link for the database (see this short tutorial).
- Upload database from the data library.
In this tutorial, we will explore using Protein Database Downloader for database search.
Using Protein Database Downloader¶
Now we get our search database. First thing we will do is to go to Tools –> Proteomics: Galaxy-P –> Get Data. Then click on Protein Database Downloader.

Since we are going to analyze a human dataset, select human database along with isoforms.
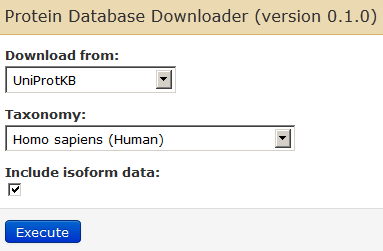
Clicking on “Execute” will download the human canonical isoform database.
After this you will see your first History Item in Galaxy’s right pane. It will go through gray (preparing) and yellow (running) states to become green:
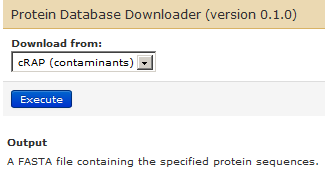
For contaminants database, we will use the Protein database Downloader, select cRAP (contaminants) database and “Execute” much the same way you did for the human UniProt database.
After this you will see your second History Item in Galaxy’s right pane.
Using the Database Merge Tool¶
In order to merge these two FASTA files in your history, go to Tools –> FASTA Manipulation –> Merge FASTA Databases.

Click on “Add new Input FASTA File(s)” – twice – so that your set up for merging files looks like this:
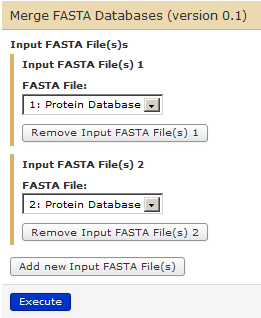
Click on Execute and you will be able to see your third History Item in Galaxy’s right pane.
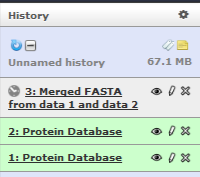
Creating a Decoy Database¶
Our next step is to create a decoy database out of the merged file (3rd history item on the list).
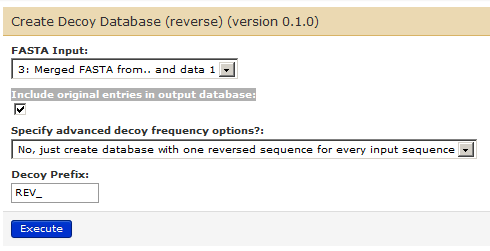
For this, go to Tools -> FASTA Manipulation –> ‘Create Decoy Database’.

Ensure that history item 3 shows up in the “FASTA Input:” box and that the box for “Include original entries in output database:” is checked. Change Decoy prefix to REV_.
Your parameters for creating decoy database (reverse) tool should look like this.
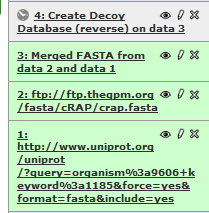
Click on “Execute” to generate the fourth item in your history list. This is the FASTA database that we will be using for our search.
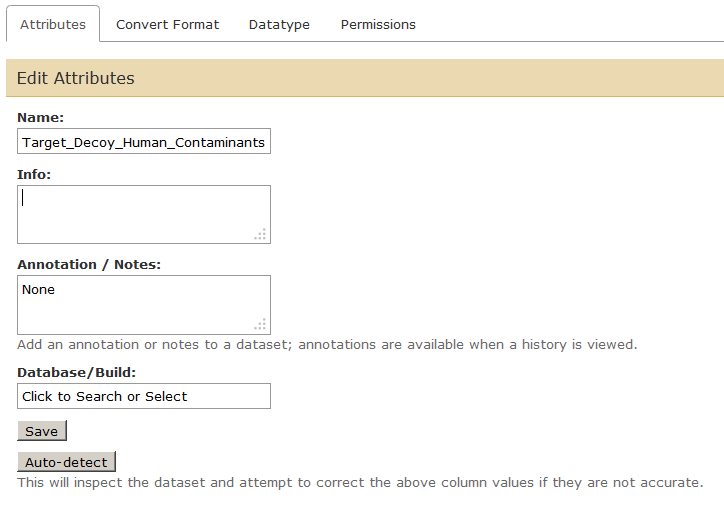
Now we will rename the history items to “Human UniProt”, “cRAP”, “Merged Human UniProt cRAP” and “Target_Decoy_Human_Contaminants” by clicking on the Pencil icon adjacent to each item. Also we will rename history to “Galaxy-P 101” (or whatever you want) by clicking on “Unnamed history” so everything looks like this:
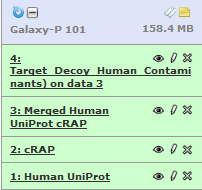
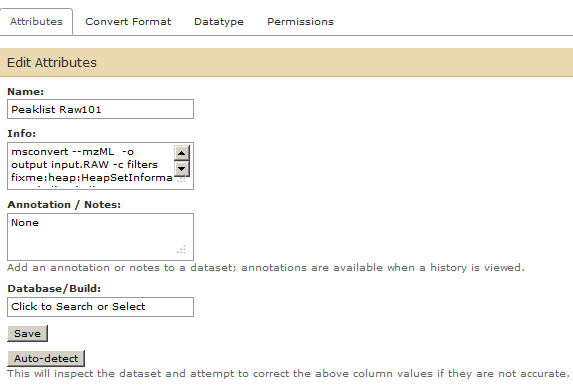
Please feel free to explore tabs – Convert Format, Datatype, Permissions while you are editing the attributes. This is especially important while troubleshooting for steps that fail wherein a datatype has not been set properly or needs to be changed for subsequent steps.
The next step is to upload RAW file.
There are a few options on how you can upload yours spectral data (RAW file acquired on a LTQ/Orbitrap) * Upload from your computer using your web browser. * Upload from your computer using the Galaxy-P FTP server. * Use website link for the RAW file (something we will use for this tutorial). * Import data from the data library.
While there are many ways of uploading a RAW file, we will use a weblink to upload a fractionated human salivary RAW file.
In order to upload a RAW file, you can go to “Upload File” in Tools section and type in https://netfiles.umn.edu/users/pjagtap/Galaxy-P 101/Raw101.RAW and “Execute”.
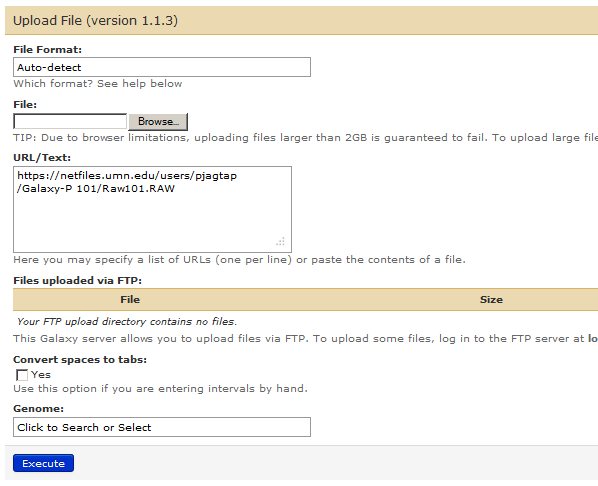
This will give us our 5th history item :
We can change the name of the RAW file to Raw101.RAW by using the pencil icon button.
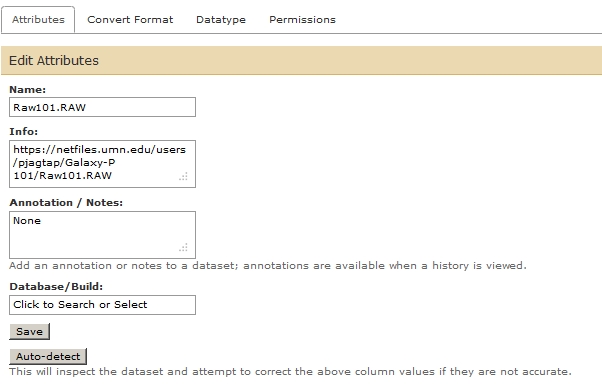
Peaklist Processing¶
In the next step we will process the Raw file into a peaklist in mzml format so that it can be searched using X!tandem.
For this go to Tools –> Proteomics: Galaxy-P –> Peak List Processing –> msconvert3 raw
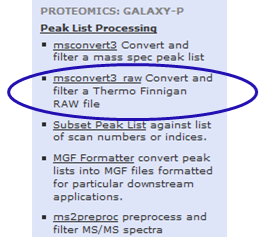
Explore the features by clicking on Use Filtering. However, for this tutorial we are going to use default settings for msconvert.
Click on “Execute” by ensuing that the settings appear as shown below:
We have our sixth history item in the history list now.

Change the name of the mzml file to “Peaklist Raw101”. Note that this peaklist is in mzml format.
X! Tandem Search¶
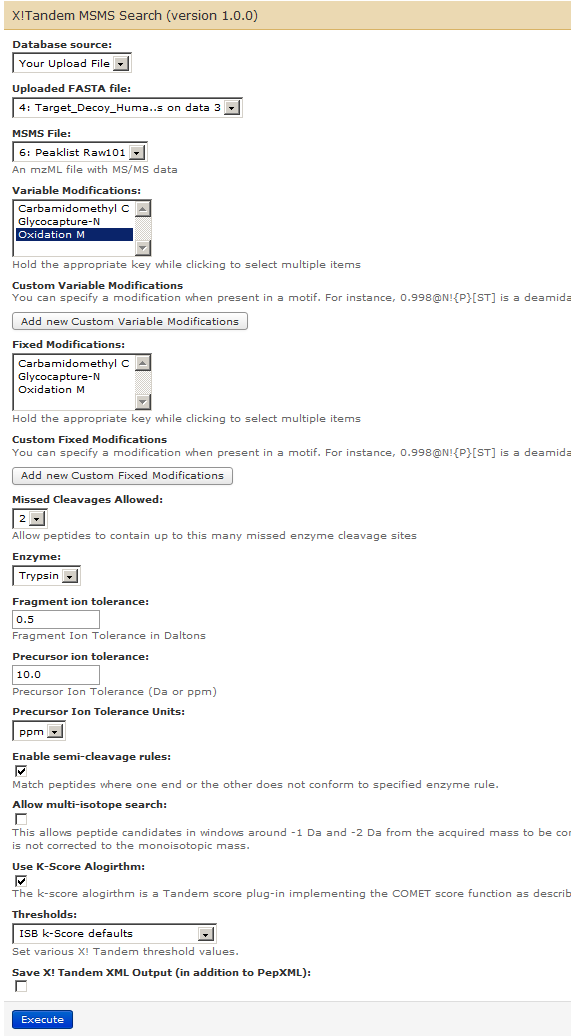
For setting up a search of peaklist (Peaklist Raw101) against FASTA database (Target_Decoy_Human_Contaminants) using X!tandem, go to Tools–> ProtK (under PROTEOMICS: COMMUNITY) –> X!Tandem MSMS Search.
Set up search parameters for X!tandem as shown below. Ensure that you have the right FASTA file (4: Target_Decoy_Human_Contaminants), MSMS File (6: Peaklist Raw101) and click on Oxidation M for variable modifications. Ensure that you have 2 Missed Cleavages allowed, Trypsin as an enzyme, Fragment ion tolerance at 0.5 Da and Precursor ion tolerance at 10.0 ppm. The parameters file should look like below:
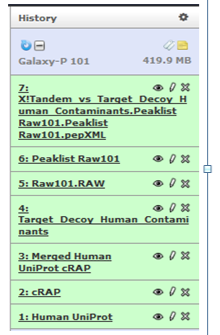
Your history list should look like this now.
Peptide Prophet Processing of X! Tandem Search¶
In this step we will process PepXML results from X! Tandem search to provide peptide probability scores for further analysis.
For this, go to Tools–> ProtK (under PROTEOMICS: COMMUNITY) –> ‘Peptide Prophet’.
The Peptide Prophet parameters should be specified as follows:

Change the name of the Peptide Prophet file to “peptide_prophet Raw101.pep.xml” by using the pencil icon.
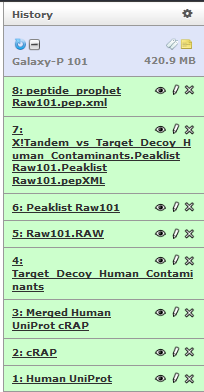
In this step, we will use ProteinProphet to process PeptideProphet results from X!tandem search to provide protein probability scores for further analysis.
For this, go to Tools –> ProtK (under PROTEOMICS: COMMUNITY)–> Protein Prophet.
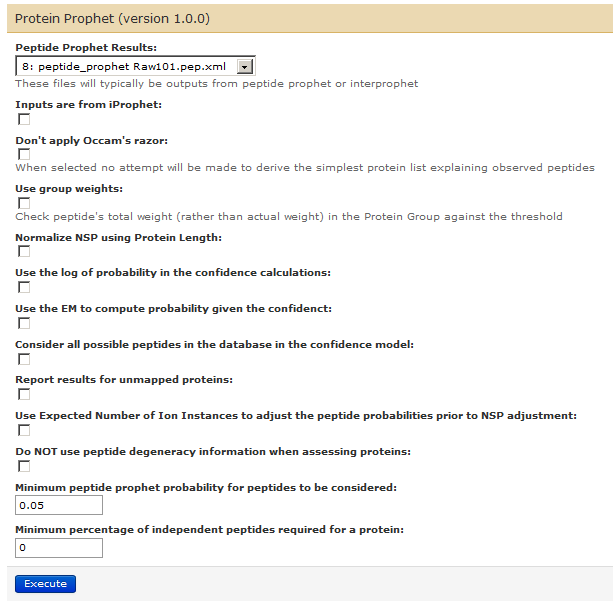
This will generate 9th history item in the list.
Converting ProtXML to a Table¶
In this step we will convert Protein Prophet results to a tabular format so that they can be viewed or processed for further analysis.
For this, go to Tools –> Utilities (under PROTEOMICS: COMMUNITY) –> Convert ProtXML to Tabular. Ensure that the latest file in the history (protXML file – 9th in the list).
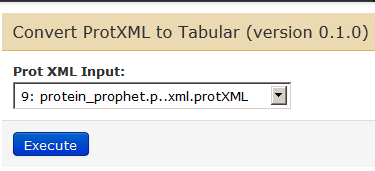
Clicking on Execute will give us our 10th file in our history.
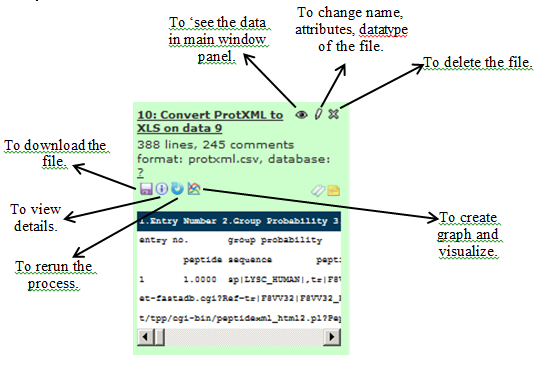
Explore the icons in the generate the file to see (‘eye’) the data OR to download (‘floppy disk’), view details (‘i” sign) OR rerun the analysis with changed parameters (blue ‘rerun’ icon). You can also create graphs and visualize using the ‘graph’ icon.
False Discovery Rate Analysis¶
In order to calculate FDR at the peptide level, we will first convert PeptideProphet file to a tabular format.
For this, go to Tools –> ProtK (under PROTEOMICS: COMMUNITY) –> Convert PepXML to Table. Ensure that the peptide prophet file in the history (peptide prophet file – 8th in the list) is highlighted.

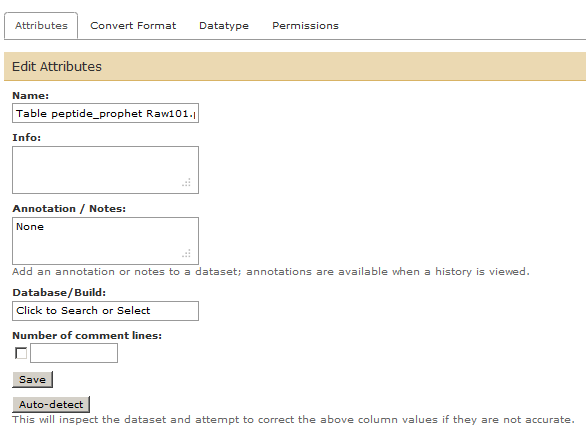
Change the name of this 11th file in history list to add “Table…” as shown in the image below.
Next step is to sort the table with descending peptide probability scores. For this go to Tools –> Filter and Sort–> Sort. Ensure that table (csv file – 11th in the list) is highlighted.
Also select c10 (10th column which has peptide probability values) for sorting in descending order.
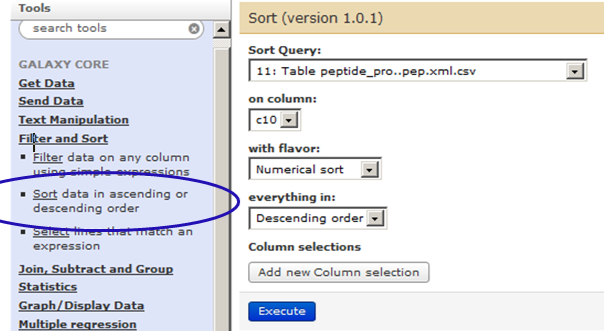
Click on ‘Execute’. This will create you 12th list in history.
To compute FDR on this file, go to Tools –> Statistical validation (under PROTEOMICS: GALAXY-P) –> Compute False discovery Rate (FDR). Ensure that the sorted table (tsv file – 12th in the list) is highlighted. Also select c1 (1st column which has identifiers) for sorting in descending order. The parameters for this processing should look as below:
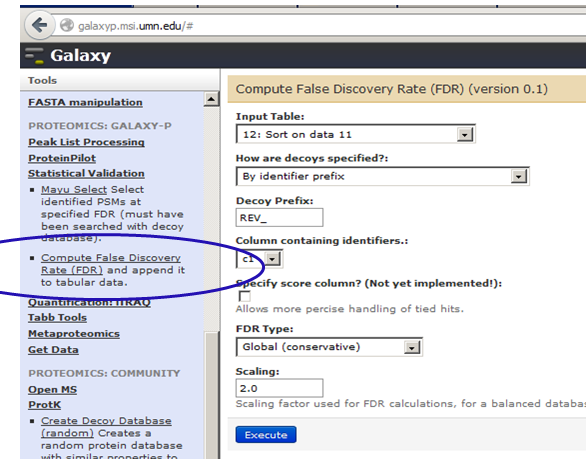
This will give you an output with a column with false discovery rate. You can use the ‘eye’ icon to visualize your data.
You can use the text manipulation tools such as ‘Add column” and “cut” in order to visualize your data better.
Go to Tools –> Text manipulation –> Add column. Ensure that the correct file (number 13 in the list) is chosen as a dataset. Change Iterate column to yes and click on execute.
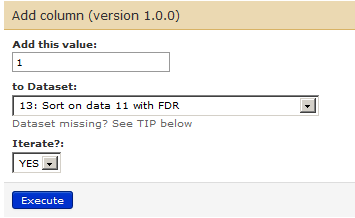
This will give you your 14th history item. Use the eye icon to confirm that a column has been added at the end of the file.
Next, we will cut columns 1, 10 12 and 13 from history dataset number 14. For this go to Tools –> Text manipulation –> Cut. Ensure that the correct file (number 14 in the list) is chosen as a dataset. Type in c1,c10,c12,c13 in ‘cut columns’ box and press execute.
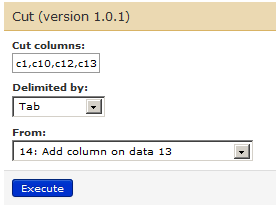
This will give you 15th history dataset.
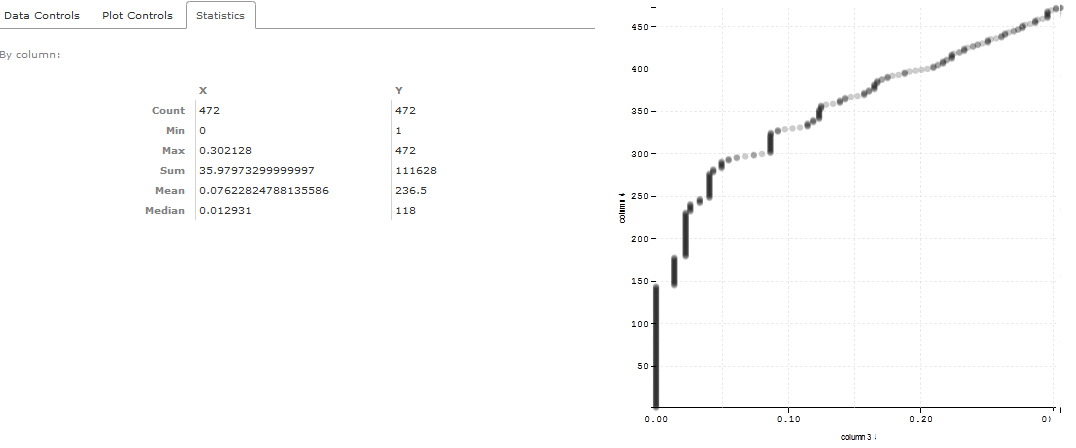
If you click on the Visualize / scatterplot icon you will see a scatterplot controls in the central window.
Keep the Data column X: column 3 and change Data column Y: column 4. Click on Draw.
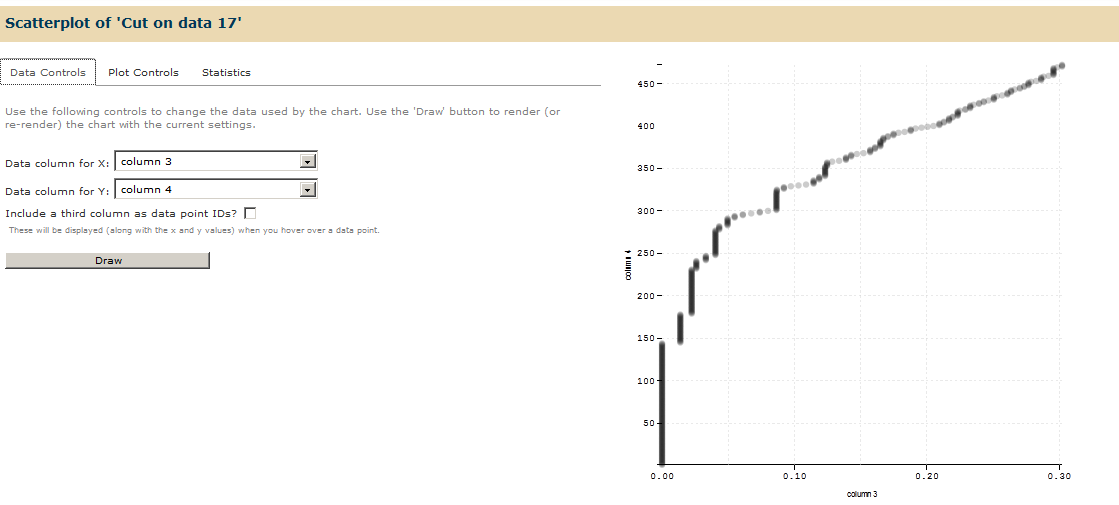
This will give you a ROC plot that gives you statistics and number of identifications at various levels of FDR.
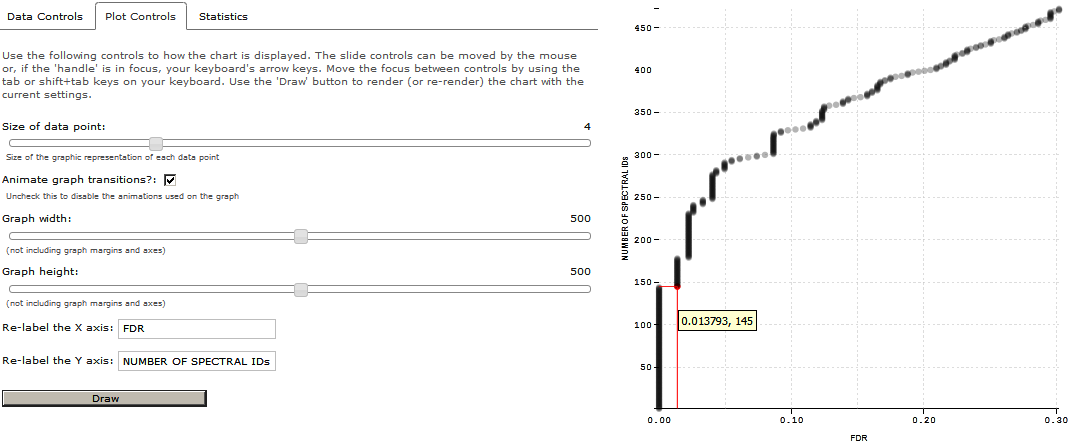
Go to “Plot Controls” Tab and change the controls on your chart. Relabel X axis as “FDR” and Y-axis as ‘NUMBER OF SPECTRAL IDs”. Click on Draw to render the chart on current settings.
If you scroll on the graph, you will be able to see that 144 spectra were identified in this dataset before the first reverse match was encountered (FDR of 1.3%). Thus, 144 spectra were identified below 1% global FDR.
Understanding Galaxy Histories¶
In Galaxy, your analyses live in histories such as this one:
Histories can be very large, you can have as many histories as you want, and all history behavior is controlled by the Options button on the top of the History pane:
Many of the options here are self explanatory. If you create a new history, your current history does not disappear. If you would like to list all of your histories just choose Saved Histories and you will see a list of all your histories in the central window pane:
Converting Histories to Workflows¶
One of the history options listed is very special. It allows you to easily convert existing histories into analysis workflows. Why would you want to create a workflows out of a history? To redo the analysis again with minimal clicking.
Lets take a look at the history again:
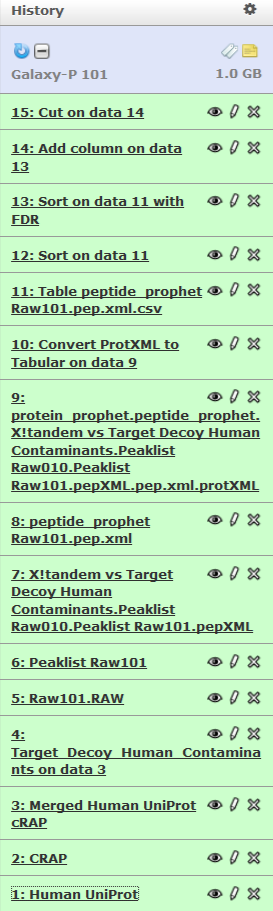
You can see that this history contains all steps of our analysis. So by building this history we have actually built a complete record of our analysis with Galaxy preserving all parameter settings applied at every step. Wouldn’t it be nice to just convert this history into a workflow that we’ll be able to execute again and again? This can be done by clicking on Options button and selecting Extract Workflow option:
The center pane will change as shown below and you will be able to choose which steps to include/exclude and how to name the newly created workflow. In this case I named it “Galaxy-P 101 Workflow”:
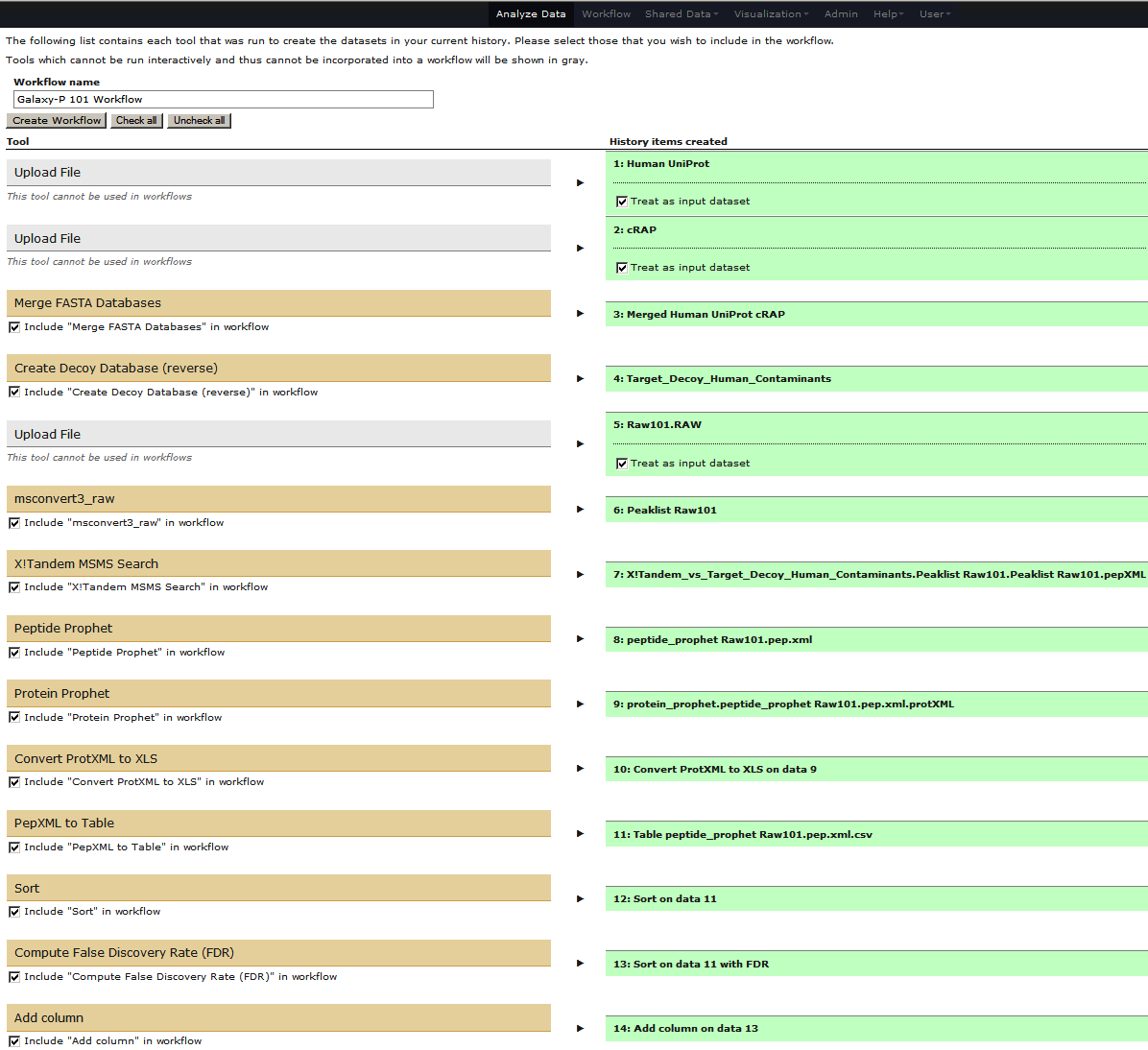
Once you click Create Workflow you will get the following message: “Workflow ‘Galaxy-P 101 Workflow’ created from current history”. But where did it go? Click on Workflow link at the top of Galaxy interface and you will a list of all workflows with “Galaxy-P 101 Workflow” listed at the top:
If you click on a triangle adjacent to the workflow’s name you will see the following dialogue:
Click Edit and the workflow editor will launch.
It will allow you to examine and change settings of this workflow as shown below. Note that you can click on each box so that you can see parameters of this tool on the right pane. This is how you can view and change parameters of all tools involved in the workflow.
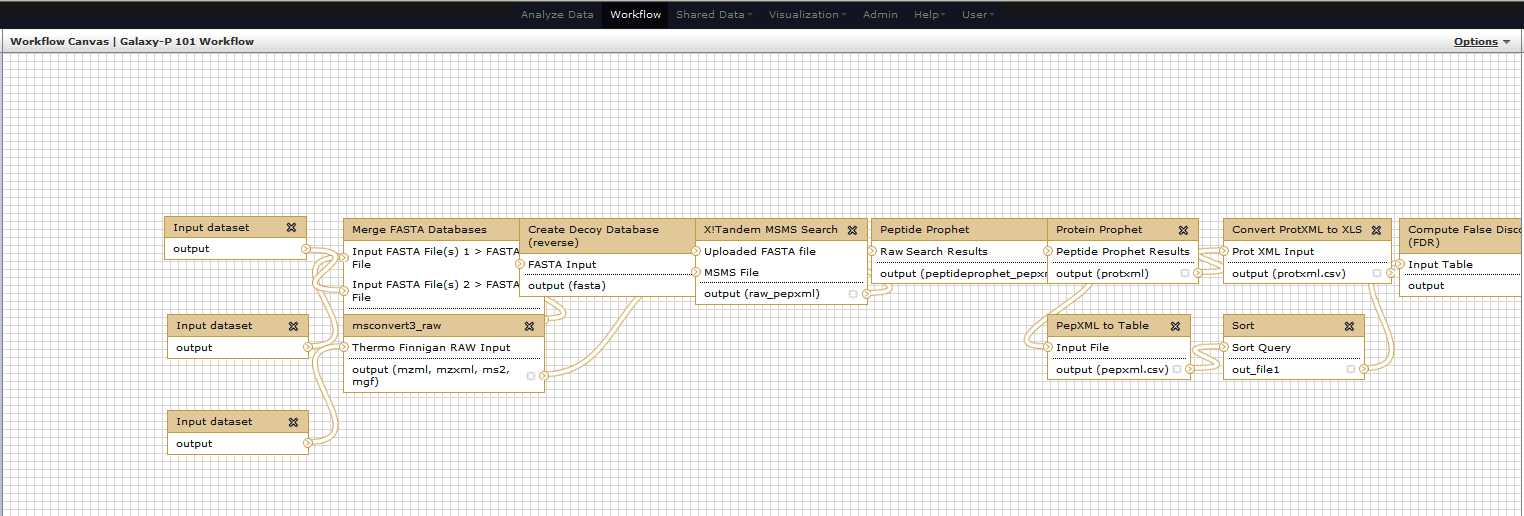
You can also reorganize your workflow so that it makes intuitive sense. For example, I have compartmentalized this workflow as follows. However, as in any form of art – this is not the only way of representing your workflow. Re- architecture as the best way you wish to.
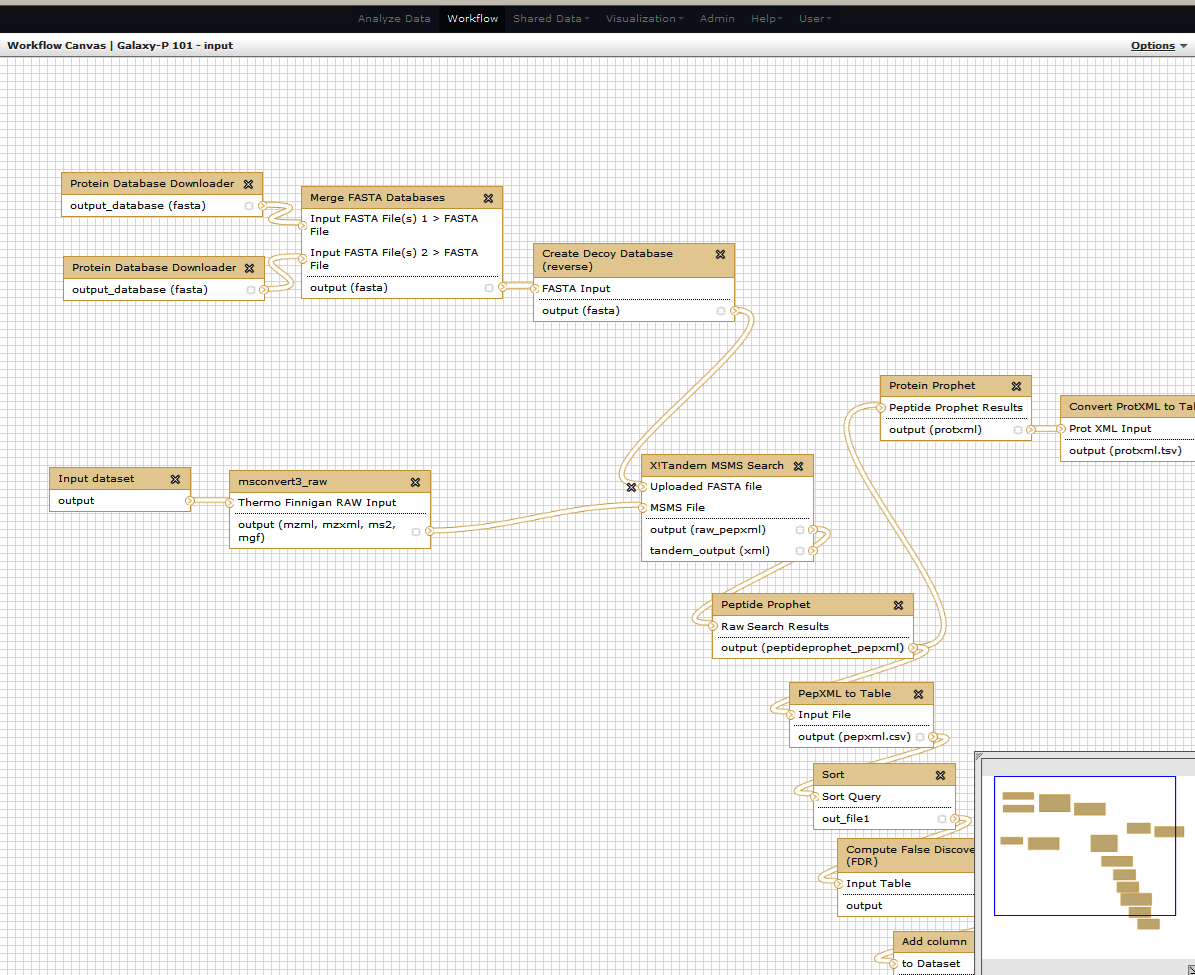
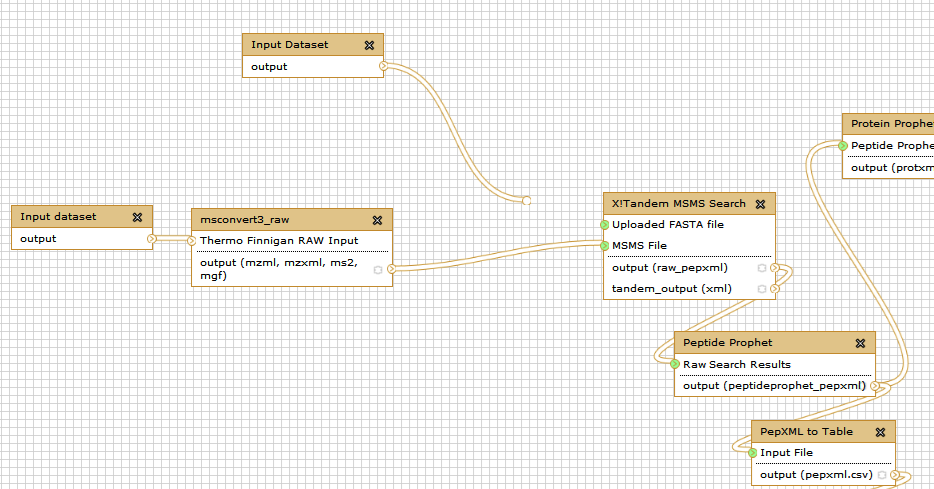
For this tutorial, we are going to reformat the workflow by replacing the earlier part of database creation with an input database. For this we excise the part where uploaded FASTA file merges with the X! Tandem search step.
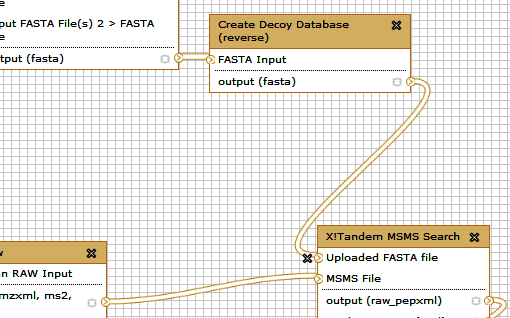
You can delete all the steps involving FASTA database creation. Delete one step at a time...
Your workflow now will look like this. Note that the Uploaded FASTA file has not input.
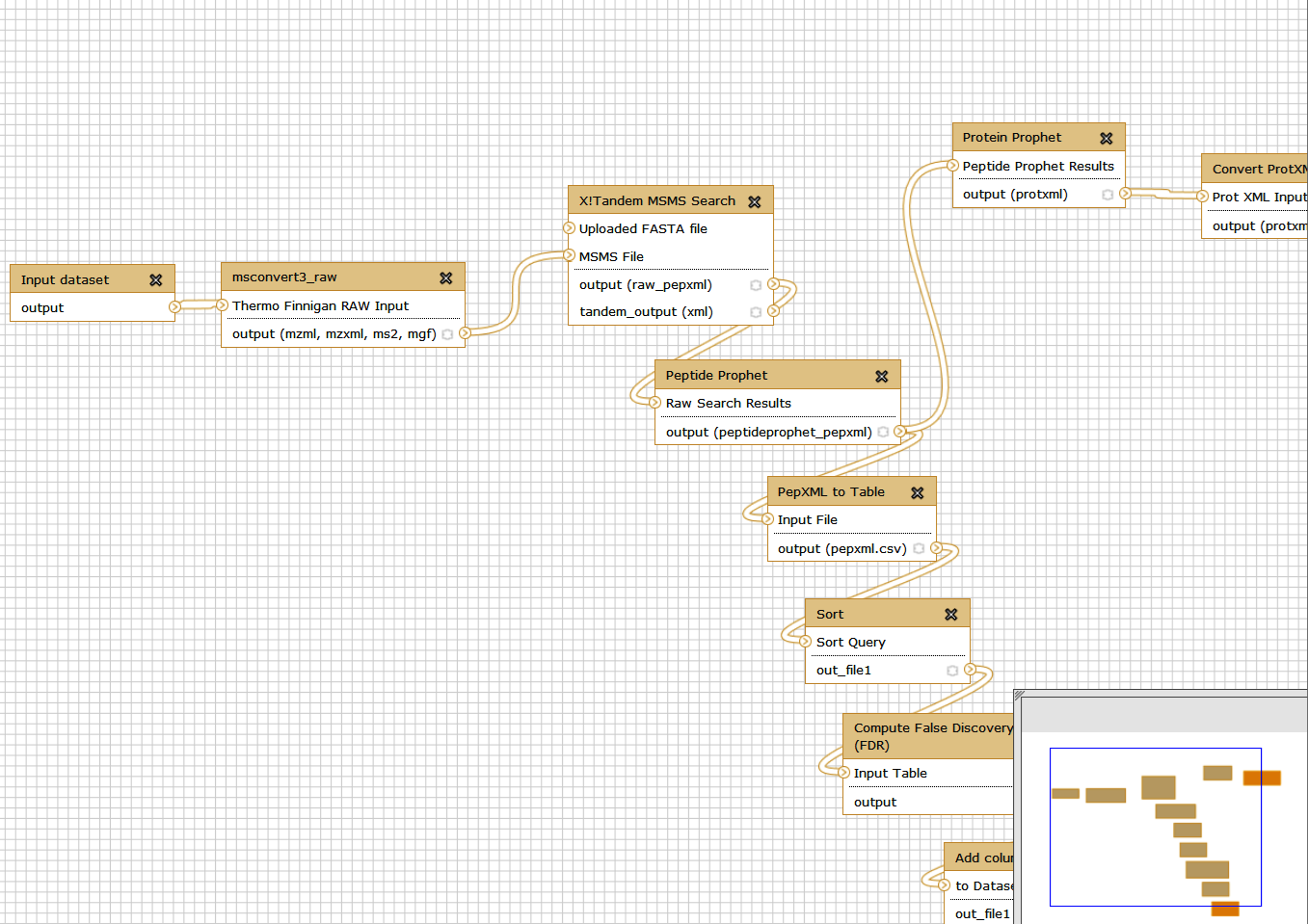
You can now scroll down to the left bottom corner of the screen and click on inputs. ‘Input dataset’ option will show up. Click on it.
Once this is done, ‘input dataset’ box will appear in the central Window pane.
Position the Input dataset pane in such a way that you can join the output arrow on this box with the input arrow for the X!tandem search. In other words, we are providing a FASTA database input for the X!tandem search. The significance of this step will become clearer when we use this workflow.
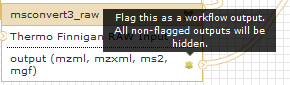
Below is mentioned one of the many things that you can do with workflows. When workflow is executed one is usually interested in the final product and not in the intermediate steps. These steps can be hidden by mousing over a small asterisk in the lower right corner of every tool box:
Yet there is a catch. In a newly created workflow all steps are hidden by default and default behavior of Galaxy is that if all steps of a given workflow are hidden, then nothing gets hidden in the history. This may be counterintuitive, but this is done to decrease the amount of clicking if you do want to hide some steps. So in our case if we want to hide all intermediate steps with the exception of the last one we will click that asterisk in last step of the workflow:
Once you do this the representation of the workflow in the bottom right corner of the editor will change with these steps becoming orange. This means that these are the only steps, which will generate datasets visible in the history:
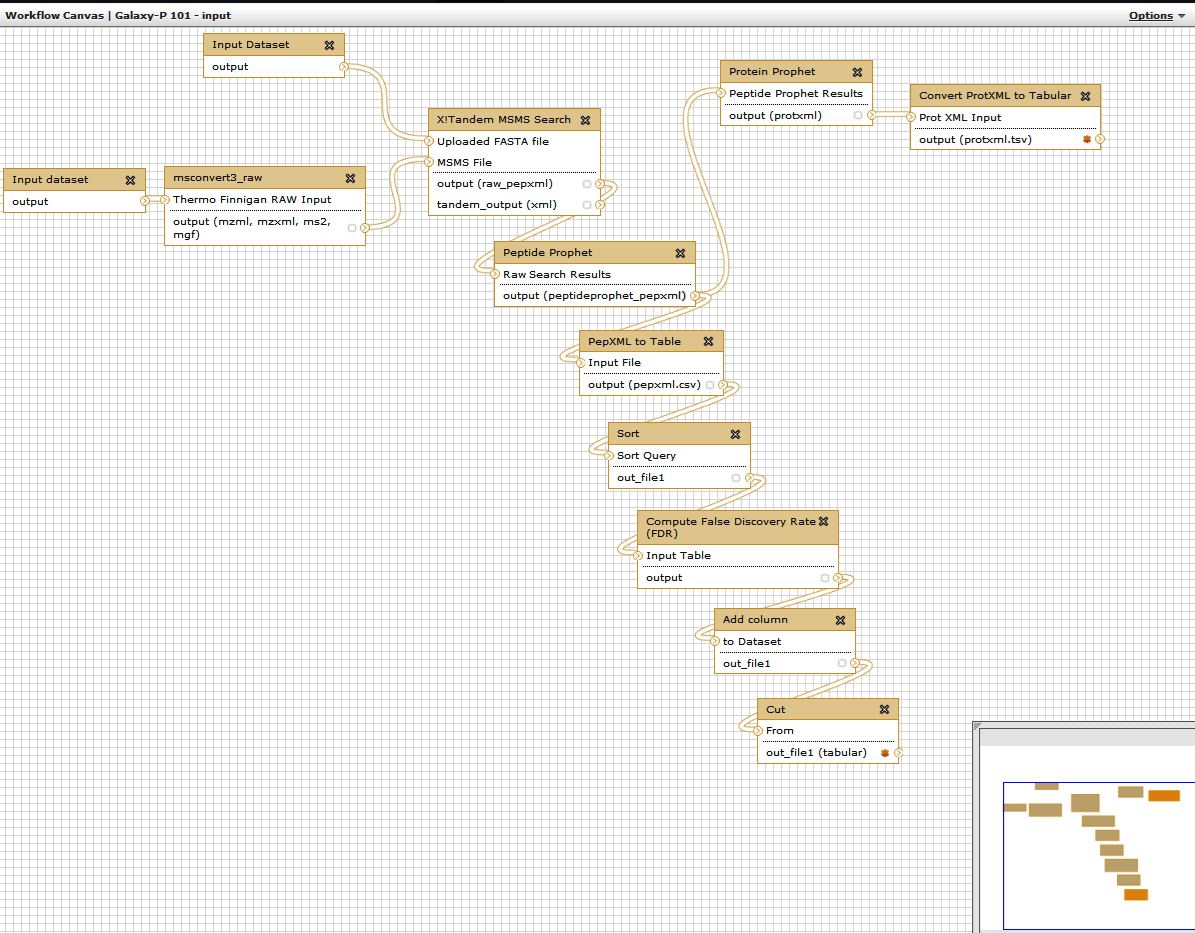
Right now both inputs to the workflow look exactly the same. This is a problem as will be very confusing which input should be FASTA files and which should be RAW file. In your workflow you will see that the top input dataset connects to the X!tandem search , so it must correspond to the Target Decoy database. If you click on this box you will be able to rename the dataset in the right pane:
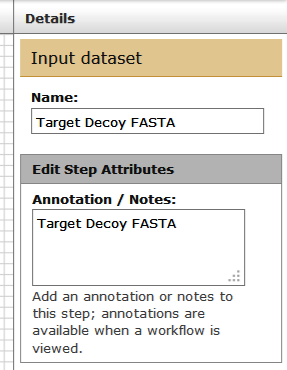
... and Thermo RAW file (second input).
Feel free to annotate as many steps as you can so that it can be easier for you to revisit and understand the workflow or easier to share it with others.
Renaming outputs
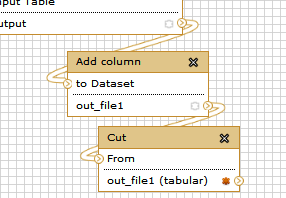
Finally let’s rename the workflow’s output. For this click on one of the last datasets (“Convert ProtXML to Tabular”) and in the Edit Step Actions dialogue box select “Rename Dataset”.
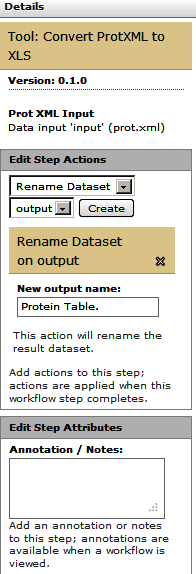
Similarly, for the other output dataset (“Cut”) , click on the box and in the Edit Step Actions dialogue box select “Rename Dataset”.
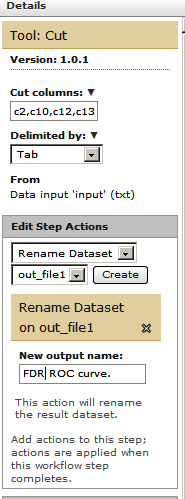
Remember you can highlight as many “outputs” as you want to and rename them for sake of a more complete and shareable annotation.
Save! It is important...
Now let’s save the changes we’ve made by clicking Options (top of the center pane) and selecting Save:
Applying Workflows to Your Data¶
Let us use this workflow on another Raw file. For this go to Analyze Data –> History options icon and select create new history.
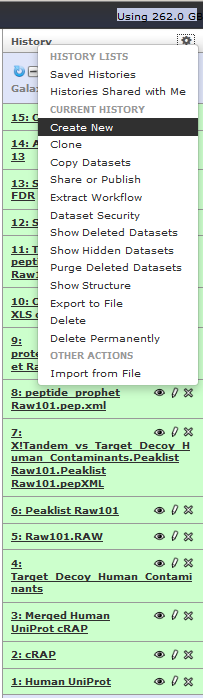
This will create a new history with no datafiles.
Let us name this history Galaxy-P 102.
We will transfer some of the files from our Galaxy-P 101 history to Galaxy-P 102 history. For this go to History options icon and click on copy datasets.
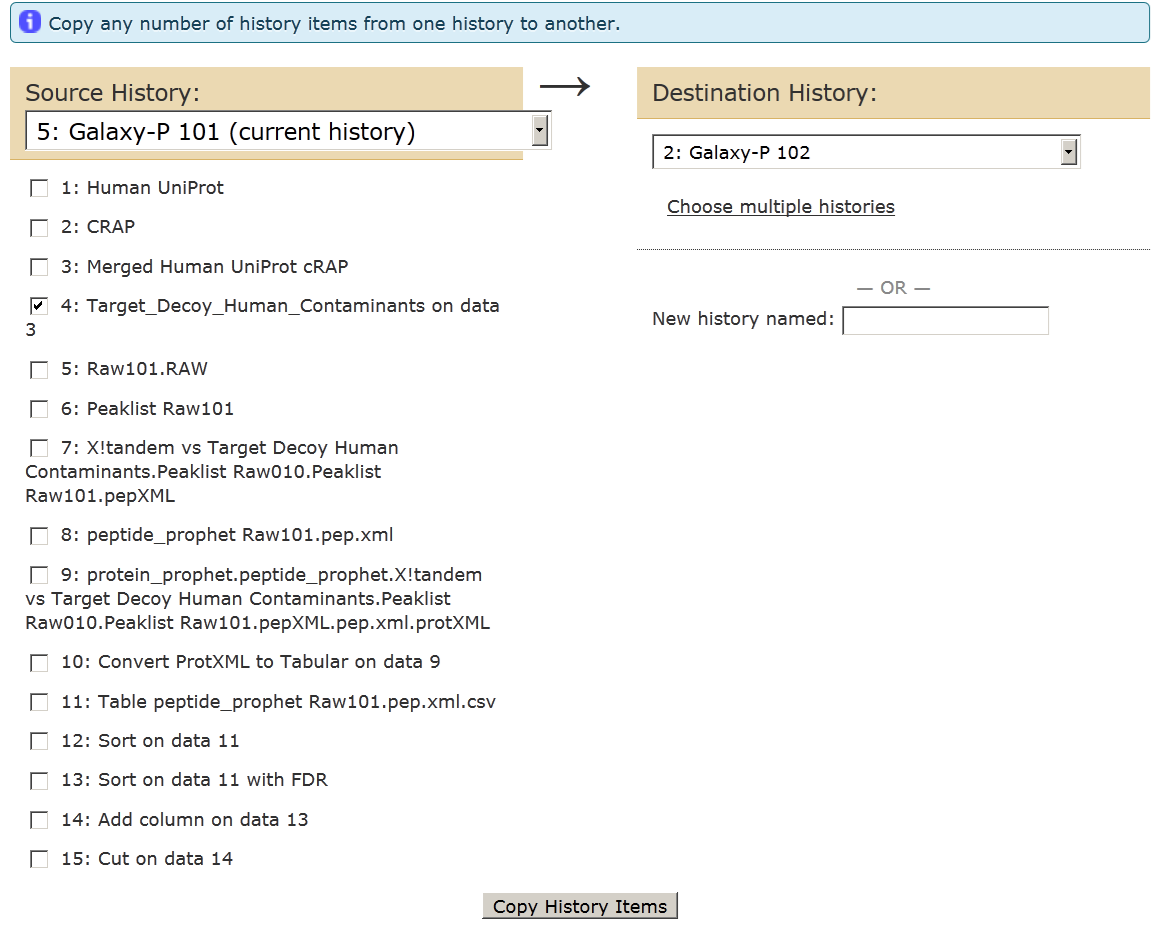
This will open up a new central window pane. Transfer the target-decoy FASTA file from Galaxy P-101 (Source History) to Galaxy-P 102 (Destination history).
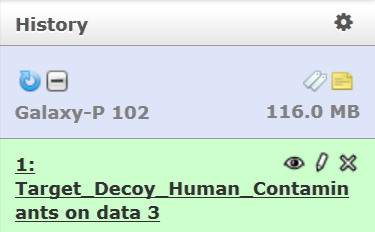
This will transfer the target-decoy FASTA file in your current history.
Click on Galaxy-P 102 history and let us download another raw file from the following link:
https://netfiles.umn.edu/users/pjagtap/Galaxy-P 101/Raw102.RAW
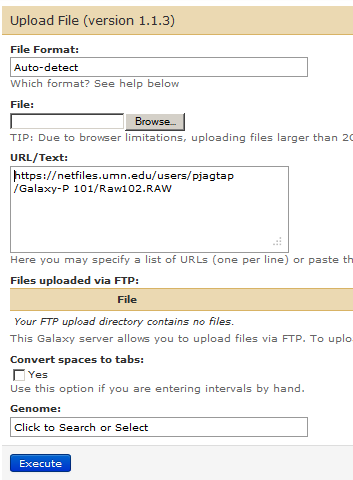
Clicking on Execute will add second file to this history.
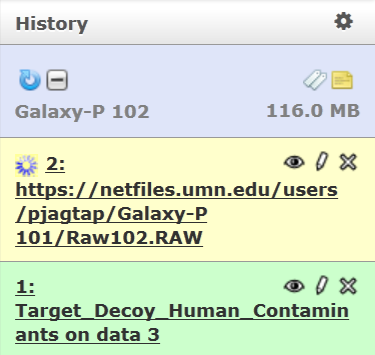
Rename the RAW file by using the pencil icon. Change the name to “Raw102”.
Now, for your rerun, your history template with 2 files (Galaxy-P 102) is ready.
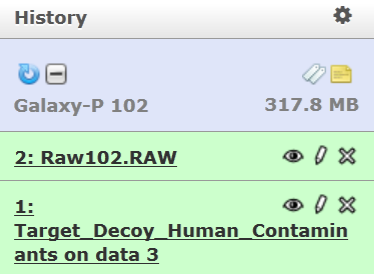
Go to Workflows tab and open the Galaxy-P 101 workflow and select “Run”.
Select appropriate files from your current history as inputs.
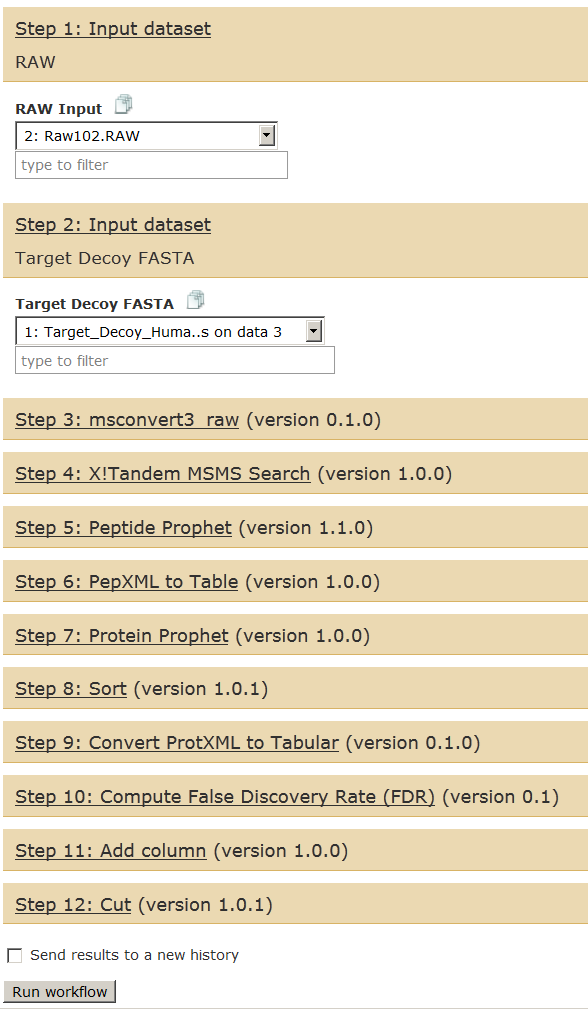
You can choose to run this in the same history or create a history of “only outputs” in a new history from this analysis. For this tutorial, we will run it in the same history.
Click on Run Workflow. Luckily you do not have to wait as Galaxy will automatically start jobs once uploads have ended.
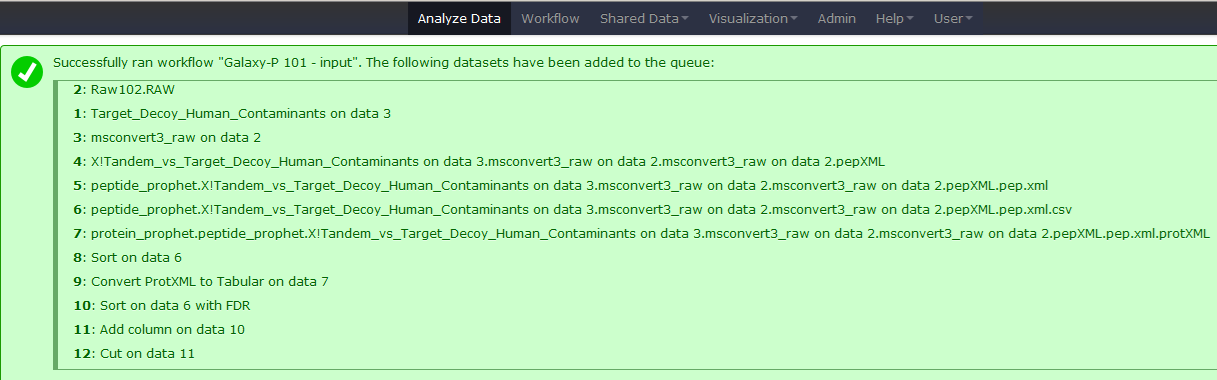
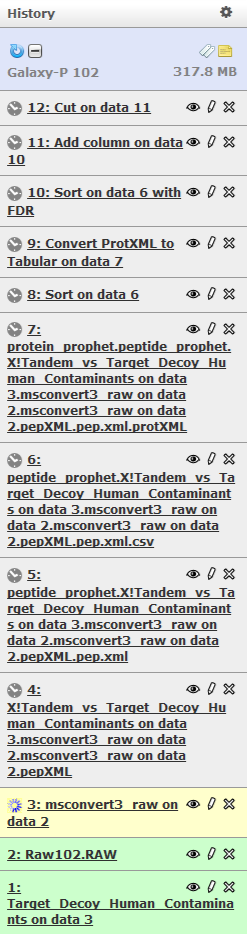
Get coffee
As we mentioned above this will take some time, so go get coffee and then you will see this. Note that because all intermediate steps of the workflow were hidden, once it is finished you will only see the final dataset.